
Noticia completa disponible en:
Nuevo antibiótico para combatir infecciones causadas por bacterias multirresistentes
NUEVO ANTIBIÓTICO PARA COMBATIR INFECCIONES CAUSADAS POR BACTERIAS MULTIRRESISTENTES
22 marzo 2024Emblaveo, de la farmacéutica Pfizer, combina dos principios activos, aztreonam y avibactam. Por un lado, aztreonam está autorizado en la UE para su uso individualizado y el avibactam está autorizado para su uso en combinación con otro antibiótico (ceftazidima).
El Comité de Medicamentos de Uso Humano (CHMP) de la EMA ha considerado que los beneficios de Emblaveo superan a los riesgos asociados para los pacientes con infecciones causadas por bacterias Gram-negativas. Emblaveo ha demostrado su eficacia en el tratamiento de diversas infecciones graves. El medicamento estará disponible en polvo concentrado para ser administrado por vía intravenosa.
Los datos de microbiología indican que la combinación de aztreonam y avibactam es efectiva en infecciones causadas por muchos patógenos Gram-negativos aerobios multirresistentes, por lo que la combinación podría responder a una necesidad médica aún no cubierta.
Los efectos secundarios más frecuentes en pacientes tratados con Emblaveo fueron anemia, aumento en los niveles de transaminasas y diarrea. Estos resultados están en consonancia con la información de seguridad disponible para cada una de las sustancias.
LA FDA APRUEBA EL PRIMER TRATAMIENTO PARA PACIENTES CON CICATRICES HEPÁTICAS DEBIDO A LA ENFERMEDAD DEL HÍGADO GRASO
14 marzo 2024La EHNA es el resultado de la progresión de la enfermedad del hígado graso no alcohólico donde la inflamación del hígado, con el tiempo, puede provocar cicatrices y disfunción del hígado. La EHNA a menudo se asocia con otros problemas de salud como presión arterial alta y diabetes tipo 2.
Rezdiffra es un activador parcial de un receptor de hormona tiroidea. La activación de este receptor por parte de este medicamento en el hígado reduce la acumulación de grasa en el hígado.
Los efectos secundarios más comunes incluyen diarrea y náuseas. El medicamento viene con ciertas advertencias y precauciones, como toxicidad en el hígado inducida por medicamentos y efectos secundarios relacionados con la vesícula biliar. Se debe evitar el uso de Rezdiffra en pacientes con cirrosis descompensada. Los pacientes deben dejar de usar el medicamento si desarrollan signos o síntomas de empeoramiento de la función hepática mientras reciben tratamiento con Rezdiffra.
El uso de Rezdiffra al mismo tiempo que otros medicamentos, en particular estatinas para reducir el colesterol, puede provocar interacciones medicamentosas potencialmente significativas.
LA FDA APRUEBA EL PRIMER MEDICAMENTO QUE AYUDA A REDUCIR LAS REACCIONES ALÉRGICAS A VARIOS ALIMENTOS TRAS UNA EXPOSICIÓN ACCIDENTAL
16 febrero 2024Xolair fue inicialmente aprobado en 2003 para tratar el asma alérgica persistente en ciertos pacientes. También está aprobado para tratar la urticaria crónica espontánea y la rinosinusitis crónica con pólipos nasales en determinados pacientes. Es el primer medicamento aprobado por la FDA para reducir las reacciones alérgicas a más de un tipo de alimento tras una exposición accidental. Es un medicamento (de la clase de los llamados anticuerpos monoclonales) que se une a la inmunoglobulina E (IgE), el tipo de anticuerpo que desencadena las reacciones alérgicas, y bloquea la unión de la IgE a sus receptores.
Los efectos secundarios más comunes de este medicamento incluyen reacciones en el lugar de la inyección y fiebre. Además, el medicamento cuenta con advertencias y precauciones, como la posibilidad de anafilaxia, malignidad y pruebas de laboratorio anormales.

Noticia completa disponible en:
https://cdn.who.int/media/docs/default-source/gcp/who-mia-list-2024-lv.pdf?sfvrsn=3320dd3d_2
OMS PUBLICA LA LISTA DE ANTIMICROBIANOS DE IMPORTANCIA PARA LA MEDICINA HUMANA
08 febrero 2024- De importancia crítica: Antimicrobianos esenciales para tratar enfermedades graves en humanos, cuyo uso en otros sectores debe ser limitado para preservar su eficacia.
- Muy importante: Antimicrobianos importantes para la medicina humana, con algunas alternativas disponibles, pero su uso debe ser gestionado cuidadosamente.
- Importante: Antimicrobianos que tienen más alternativas en medicina humana, pero que se debe gestionar su uso para evitar el aumento de la RAM.

LA AGENCIA EUROPEA DE MEDICAMENTOS (EMA) HA CONFIRMADO MEDIDAS PARA MINIMIZAR EL RIESGO DE EFECTOS SECUNDARIOS GRAVES EN MEDICAMENTOS QUE CONTIENEN PSEUDOEFEDRINA
26 enero 2024- Síndrome de encefalopatía posterior reversible (PRES): Implica una reducción del flujo sanguíneo al cerebro, lo que puede causar complicaciones serias y potencialmente mortales.
- Síndrome de vasoconstricción cerebral reversible (RCVS): También afecta el flujo sanguíneo cerebral y puede tener consecuencias graves.
Estas recomendaciones se basan en una revisión exhaustiva de la evidencia disponible y se aplicarán a todos los medicamentos que contienen pseudoefedrina. La opinión del CHMP se enviará a la Comisión Europea, que tomará una decisión legalmente vinculante en toda la Unión Europea
Noticia completa en:
https://www.prescrire.org/Fr/202/1834/55640/0/PositionDetails.aspx
ACTUALIZACIÓN DE LOS MEDICAMENTOS A EVITAR SEGÚN LA REVISTA PRESCRIRE
05 enero 2024- Sustancias activas con efectos adversos que, dadas las situaciones clínicas en las que se utilizan, son desproporcionados con respecto a los beneficios que aportan
- Medicamentos más antiguos que han sido reemplazados por medicamentos más nuevos con mejor relación riesgo-beneficio
- Medicamentos recientes que tienen una relación riesgo-beneficio menos favorable que otras opciones
- Medicamentos que no tienen eficacia probada más allá del efecto placebo, pero que conllevan un riesgo de efectos adversos particularmente graves

LA FDA APRUEBA LAS PRIMERAS TERAPIAS GÉNICAS PARA TRATAR A PACIENTES CON ANEMIA DE CÉLULAS FALCIFORMES
08 diciembre 2023La anemia de células falciformes es un grupo de trastornos sanguíneos hereditarios que afectan aproximadamente a 100,000 personas en los Estados Unidos. Es más común en afroamericanos y, aunque menos prevalente, también afecta a hispanoamericanos. El problema principal de la anemia de células falciformes es una mutación en la hemoglobina, una proteína que se encuentra en los glóbulos rojos y que suministra oxígeno a los tejidos del cuerpo. Esta mutación hace que los glóbulos rojos desarrollen una forma de media luna o de “hoz.” Estos glóbulos rojos falciformes restringen el flujo en los vasos sanguíneos y limitan el suministro de oxígeno a los tejidos del cuerpo, lo que provoca un dolor intenso y daño a los órganos, conocidos como episodios vasooclusivos (VOE, por sus siglas en inglés) o crisis vasooclusivas (VOC, por sus siglas en inglés). La recurrencia de estos eventos o crisis puede provocar discapacidades potencialmente mortales o muerte prematura.
LA FDA APRUEBA LA PRIMERA VACUNA PARA PREVENIR LA ENFERMEDAD CAUSADA POR EL VIRUS DEL CHIKUNGUNYA
09 noviembre 2023El virus del chikungunya se transmite principalmente a las personas a través de la picadura de un mosquito infectado. El chikungunya es una amenaza emergente para la salud mundial, con al menos 5 millones de casos de infección por el virus del chikungunya reportados durante los últimos 15 años. El mayor riesgo de infección se encuentra en las regiones tropicales y subtropicales de África, el sudeste asiático y partes de América donde los mosquitos portadores del virus del chikungunya son endémicos. Sin embargo, el virus del chikungunya se ha propagado a nuevas áreas geográficas provocando un aumento en la prevalencia global de la enfermedad.
Los síntomas más comunes del chikungunya incluyen fiebre y dolor en las articulaciones. Otros síntomas pueden incluir sarpullido, dolor de cabeza y dolor muscular. Algunas personas pueden experimentar un dolor articular debilitante que persiste durante meses o incluso años. El tratamiento incluye reposo, líquidos y medicamentos de venta libre para el dolor y la fiebre.

OCHO NUEVOS MEDICAMENTOS RECOMENDADOS PARA SU APROBACIÓN
10 noviembre 2023Después de un nuevo examen , el comité recomendó conceder autorización de comercialización condicional a Krazati (adagrasib), para el tratamiento de adultos con cáncer de pulmón de células no pequeñas avanzado con una mutación G12C en el gen KRAS.

FLUOROQUINOLONAS DE ADMINISTRACIÓN SISTÉMICA O INHALADA: RECORDATORIO SOBRE LAS RESTRICCIONES DE USO
23 octubre 2023Los resultados del estudio EUPAS37856 realizado entre 2016 y 2021 en seis bases de datos europeas, sugieren que estos medicamentos podrían seguir prescribiéndose fuera de las indicaciones autorizadas, a pesar de que su uso se ha reducido en el último período. Los resultados no son concluyentes.
La Agencia Española de Medicamentos y Productos Sanitarios, en colaboración con el Plan Nacional frente a la Resistencia a los Antibióticos, realizó una encuesta para evaluar el nivel de conocimiento y comprensión de los mensajes clave de seguridad relacionados con las restricciones de uso de fluoroquinolonas de administración sistémica o inhalada en profesionales sanitarios. Los resultados de la encuesta muestran que la mayoría de los profesionales de la salud respondieron positivamente.
NUEVA OPCIÓN DE TRATAMIENTO PARA PACIENTES CON MIELOMA MÚLTIPLE TRATADOS PREVIAMENTE
13 octubre 2023El ranatamab es el principio activo de Elrexfio, es un anticuerpo monoclonal que se dirige a dos proteínas simultáneamente. Este medicamento recibió apoyo a través del esquema PRIority MEdicines (PRIME) de la EMA.
SPIKEVAX: LA EMA RECOMIENDA LA APROBACIÓN DE LA VACUNA COVID-19 ADAPTADA DIRIGIDA A OMICRON XBB.1.5
14 septiembre 2023El comité de medicamentos humanos (CHMP) de la EMA ha recomendado autorizar una vacuna Spikevax adaptada dirigida a la subvariante Omicron XBB.1.5.
La vacuna, conocida como Spikevax XBB.1.5, se utilizará para prevenir la COVID-19 en adultos y niños a partir de los 6 meses de edad.
De acuerdo con recomendaciones anteriores de la EMA y el Centro Europeo para la Prevención y el Control de Enfermedades (ECDC), los adultos y niños a partir de 5 años que requieran vacunación deben recibir una dosis única, independientemente de su historial de vacunación contra la COVID-19. Los niños de 6 meses a 4 años podrán recibir una o dos dosis dependiendo de si han completado un ciclo de primovacunación o han tenido COVID-19.
En su decisión de recomendar la autorización, el CHMP consideró todos los datos disponibles sobre Spikevax y sus otras vacunas adaptadas. Además, el comité evaluó datos de laboratorio que muestran que la vacuna adaptada es capaz de desencadenar una respuesta inmune adecuada contra XBB.1.5.
El CHMP también consideró datos de un estudio en el que se administró Spikevax XBB.1.5 a adultos como refuerzo. El estudio demostró que la vacuna produjo una respuesta inmune contra la subvariante Omicron XBB.1.5, medida por un aumento en el nivel de anticuerpos contra esta cepa. La vacuna también produjo una respuesta inmune contra otras cepas del virus que causa el COVID19, incluida la subvariante Omicron XBB.1.16 que circula actualmente.
La EMA enviará ahora la recomendación del CHMP a la Comisión Europea para una decisión jurídicamente vinculante en toda la Unión Europea.

LA FDA EMPRENDE ACCIONES CON RESPECTO A LAS VACUNAS CONTRA EL COVID-19 DE ARNM ACTUALIZADAS PARA MEJORAR LA PROTECCIÓN CONTRA LAS VARIANTES QUE CIRCULAN ACTUALMENTE
11 septiembre 2023Hoy, la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA, por sus siglas en inglés) tomó medidas para aprobar y autorizar el uso de emergencia de vacunas contra el COVID-19 actualizadas, formuladas para atacar más de cerca a las variantes que circulan actualmente y brindar una mejor protección contra las consecuencias graves del COVID-19, incluyendo la hospitalización y la muerte. Las acciones de hoy se refieren a las vacunas de ARNm actualizadas para el 2023-2024 fabricadas por ModernaTX Inc. y Pfizer Inc. De acuerdo con la totalidad de la evidencia y la revisión de asesores expertos de la FDA, estas vacunas se han actualizado para incluir un componente monovalente (único) que corresponde a la variante Ómicron XBB.1.5.
LA FDA APRUEBA EL PRIMER PRODUCTO BIOSIMILAR PARA TRATAR LA ESCLEROSIS MÚLTIPLE
18 agosto 2023
LA FDA APRUEBA EL PRIMER TRATAMIENTO ORAL PARA LA DEPRESIÓN POSPARTO
04 agosto 2023La Administración de Medicamentos y Alimentos (FDA) aprobó Zuranolona (Zurzuvae) el primer medicamento oral para el tratamiento de la depresión posparto (DPP).
La DPP se caracteriza por la tristeza o la pérdida de interés en actividades que antes se disfrutaban y una disminución de la capacidad de sentir placer. Puede presentarse con síntomas como deterioro cognitivo, sentimientos de tristeza o incompetencia, pérdida de energía o pensamientos suicidas.
La FDA indicó que los efectos secundarios más comunes incluyen somnolencia, mareos, diarrea, fatiga, nasofaringitis (el resfriado común) e infección del tracto urinario. La agencia también indicó que el uso de la droga puede causar pensamientos y conductas suicidas. También puede causar daño fetal. Las mujeres deben usar métodos anticonceptivos efectivos mientras toman el medicamento y durante una semana después de tomarlo.
La dosis diaria recomendada de Zurzuvae es de 50 mg. Debe tomarse una vez al día, durante 14 días, por la noche con una comida grasa.

LA FDA FDA APRUEBA UN NUEVO MEDICAMENTO PARA PREVENIR EL VIRUS RESPIRATORIO SINCITIAL EN BEBÉS Y NIÑOS PEQUEÑOS
17 julio 2023
La Administración de Alimentos y Medicamentos (FDA) de EE.UU. aprobó Beyfortus (Nirsevimab-alip) para prevenir la enfermedad respiratoria producida por el virus sincicial respiratorio (VSR) en neonatos, bebés y niños pequeños.
La FDA otorgó esta aprobación a AstraZeneca, este medicamento ayudará a la prevención de bebés o niños que se enfrenten a la primera circulación del virus, y también a aquellos de hasta dos años que sigan siendo vulnerables a la enfermedad.
La enfermedad se contagia de persona a persona por el contacto con alguien infectado.
El virus causa una infección respiratoria aguda en personas de todas las edades y, si bien la mayoría de los infantes y niños experimenta síntomas leves, como los de un resfrío, algunos infantes, especialmente en su primera infección, desarrollan enfermedades como neumonía y bronquitis.
Beyfortus es un anticuerpo monoclonal con actividad contra el VSR. La seguridad y eficacia de Beyfortus fueron respaldadas por tres ensayos clínicos.

LA FDA APRUEBA EL PRIMER TRATAMIENTO PARA EL ESTREÑIMIENTO FUNCIONAL PEDIÁTRICO 12 junio 2023
La FDA ha aprobado Linzess (linaclotida) cápsulas para tratar el estreñimiento funcional en pacientes pediátricos de 6 a 17 años de edad. Linzess es el primer tratamiento para el estreñimiento funcional pediátrico. La dosis recomendada en pacientes pediátricos de 6 a 17 años es de 72 mcg por vía oral una vez al día.
El estreñimiento funcional es una afección frecuente en niños y adolescentes en la que los pacientes tienen deposiciones poco frecuentes con heces duras que pueden ser difíciles o dolorosas de expulsar. No se conoce una causa orgánica subyacente y, por lo general, existen múltiples factores contribuyentes.

ESTRATEGIA, VIABILIDAD Y SOSTENIBILIDAD, EJES PARA AUMENTAR PRODUCCIÓN DE PRODUCTOS MÉDICOS Y VACUNAS EN LAS AMÉRICAS: DIRECTOR DE OPS
7 junio 2023
Jarbas Barbosa, director de la OPS, afirmó que asegurar el acceso equitativo a vacunas, medicamentos y tecnologías sanitarias es una de las lecciones de la pandemia y una de las prioridades de su gestión, la que comenzó en febrero de este año.
“En América Latina y el Caribe casi el 90% de los productos médicos son importados”, detalló el directivo, y destacó la importancia de “reducir esta dependencia a las importaciones y la vulnerabilidad” que trae aparejada, por las fluctuaciones que puede haber en la cadena de suministro mundial.
El Dr. Barbosa considera que “uno de los principales errores que experimentamos como sistema de salud pública durante la pandemia fue que no fuimos capaces de garantizar un acceso equitativo a las vacunas, a los respiradores, a los medicamentos, y a los equipos de protección personal, como mascarillas y guantes. Por eso, debemos estar mejor preparados para la próxima pandemia”.
Además, consideró que el camino a seguir es aumentar la capacidad de producción regional, algo que puede lograrse con proyectos que sean “estratégicos, viables y sostenibles”. Y destacó el establecimiento en 2022 de la Plataforma Regional sobre Acceso e Innovación para Tecnologías Sanitarias.

EFICACIA Y SEGURIDAD DEL TRATAMIENTO CON MOLNUPIRAVIR PAR COVID-19: REVISIÓN SISTEMÁTICA Y METAANÁLISIS 26 mayo 2023
Métodos Se realizaron búsquedas en PubMed, Embase, CENTRAL (Registro Cochrane Central de Ensayos Controlados), ClinicalTrials.gov, ICTRP (International Clinical Trials Registry Platform) y medRxiv para identificar ensayos controlados aleatorios (ECA) relevantes desde su inicio hasta el 1 de enero de 2023. Se utilizó la herramienta Cochrane de riesgo de sesgo para ensayos aleatorios con el fin de evaluar el riesgo de sesgo de los estudios incluidos. Se utilizó el programa informático Revman 5.4 para el metanálisis.
Resultados Se incluyeron nueve ECA, con 31 573 pacientes de COVID-19, de los cuales 15 846 recibieron molnupiravir. Los resultados del metanálisis mostraron que el grupo de molnupiravir presentaba una mayor proporción en términos de mejoría clínica (RR del día 5: 2,41; IC del 95%: 1,18-4,92; RR del día 10: 1,45; IC del 95%: 1,04-2,01) y de negatividad de la reacción en cadena de la polimerasa en tiempo real (RR del día 5: 2,78; IC del 95%: 1,38-5,62; RR del día 10: 1,18; IC del 95%: 1,07-1,31). Sin embargo, no se observaron diferencias significativas entre los dos grupos en cuanto a mortalidad, hospitalización, acontecimientos adversos y acontecimientos adversos graves.
Conclusiones Molnupiravir puede acelerar la rehabilitación de los pacientes con COVID-19, pero no reduce significativamente la mortalidad ni la hospitalización
ANTIBIÓTICOS DE FLUOROQUINOLONA: RECORDATORIO DE LAS MEDIDAS PARA REDUCIR EL RIESGO DE EFECTOS SECUNDARIOS DURADEROS, INCAPACITANTES Y POTENCIALMENTE IRREVERSIBLES
12 mayo 2023
Estas restricciones se introdujeron en 2019 tras una revisión en toda la UE de estos efectos secundarios muy poco frecuentes, pero graves. Un estudio financiado por la EMA ha demostrado que, aunque se ha reducido el uso de antibióticos fluoroquinolónicos, estos medicamentos aún pueden prescribirse fuera de sus usos recomendados.
Las restricciones en el uso de antibióticos fluoroquinolónicos significan que no deben utilizarse para tratar infecciones que podrían mejorar sin tratamiento:
- Para tratar infecciones que podrían mejorar sin tratamiento o que no son graves (como las infecciones de garganta);
- Para tratar infecciones no bacterianas, por ejemplo, prostatitis no bacteriana (crónica);
- Para prevenir la diarrea del viajero o las infecciones recurrentes del tracto urinario inferior (infecciones de orina que no se extienden más allá de la vejiga);
- Para tratar infecciones bacterianas de leves a moderadas, a menos que se utilicen habitualmente otros medicamentos antibacterianos.
LA FDA AUTORIZA CAMBIOS PARA SIMPLIFICAR EL USO DE VACUNAS BIVALENTES DE ARNM CONTRA EL COVID-19
18 Abril 2023
La Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) enmendó las autorizaciones de uso de emergencia de las vacunas bivalentes de ARNm contra el COVID-19 Moderna y Pfizer-BioNTech para simplificar el esquema de vacunación para la mayoría de las personas. Esta acción incluye la autorización del uso de las vacunas bivalentes actuales (cepas originales y ómicron BA.4/BA.5) para todas las dosis administradas a personas de 6 meses de edad o mayores, incluso para una o varias dosis adicionales para ciertas poblaciones. Las vacunas monovalentes contra el COVID-19 de Moderna y de Pfizer-BioNTech ya no están autorizadas para su uso en los Estados Unidos.
EMA RECOMIENDA LA APROBACIÓN DE BIMERVAX COMO VACUNA DE REFUERZO CONTRA EL COVID-19
30 Marzo 2023
El comité de medicamentos humanos (CHMP) de la EMA ha recomendado autorizar la vacuna COVID-19 Bimervax (anteriormente COVID-19 Vaccine HIPRA) como refuerzo en personas mayores de 16 años que hayan sido vacunadas con una vacuna de ARNm COVID-19.
Bimervax, desarrollado por HIPRA Human Health S.L.U., contiene una proteína producida en laboratorio que consiste en parte de la proteína del pico del SARS-CoV-2 de las variantes del virus Alfa y Beta.
El CHMP concluyó que ya se dispone de datos suficientemente sólidos sobre la calidad, la seguridad y la inmunogenicidad de la vacuna para recomendar su autorización de comercialización en la UE.
El estudio principal realizado con Bimervax es un ensayo de inmunopuente, que comparó la respuesta inmunitaria desencadenada por esta nueva vacuna con la desencadenada por la vacuna de ARNm autorizada Comirnaty que se dirige a la proteína de pico original (Wuhan) SARS-CoV-2.
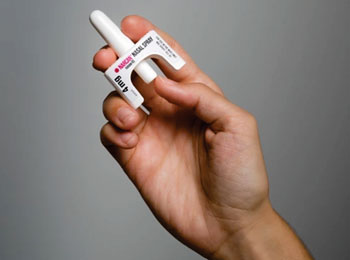
LA FDA APRUEBA EL PRIMER AEROSOL NASAL DE NALOXONA DE VENTA LIBRE
29 de Marzo de 2023
Hoy, la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA, por sus siglas en inglés) aprobó Narcan, un aerosol nasal de clorhidrato de naloxona de 4 miligramos (mg), para uso de venta libre, sin receta médica; es el primer producto de naloxona aprobado para su uso sin prescripción. La naloxona es un medicamento que revierte rápidamente los efectos de una sobredosis de opioides y es el tratamiento acostumbrado para la sobredosis de opioides. La acción de hoy cimienta el camino para que este medicamento que salva vidas revierta una sobredosis de opioides y se venda directamente a los consumidores en lugares como farmacias, tiendas de conveniencia, supermercados y gasolineras, así como en línea.
El plazo para la disponibilidad y el precio de este producto de venta libre lo determina el fabricante. La FDA trabajará con todas las partes interesadas para ayudar a facilitar la disponibilidad constante de productos de aerosol nasal de naloxona durante el tiempo necesario para efectuar el cambio de que Narcan pase a ser un medicamento que requiere receta médica a uno de venta libre, lo que puede tardar meses. Otras formulaciones y dosis de naloxona permanecerán disponibles solo con receta médica.

LA OMS A TRAVÉS DE SAGE ACTUALIZA LA GUÍA DE VACUNACIÓN COVID-19
24 de marzo de 2023
Tras su reunión del 20 al 23 de marzo, el Grupo de Expertos en Asesoramiento Estratégico sobre Inmunización (SAGE) de la OMS revisó la hoja de ruta para priorizar el uso de las vacunas COVID-19, para reflejar el impacto de Omicron y el alto nivel de inmunidad de la población debido a la infección y la vacunación.
La hoja de ruta continúa con la priorización de SAGE de proteger a las poblaciones con mayor riesgo de muerte y enfermedades graves por la infección por SARS-CoV-2 y su enfoque en mantener sistemas de salud resilientes. La hoja de ruta considera nuevamente la rentabilidad de la vacunación contra el COVID-19 para aquellos con menor riesgo, es decir, niños y adolescentes sanos, en comparación con otras intervenciones de salud. La hoja de ruta también incluye recomendaciones revisadas sobre dosis de refuerzo adicionales y el espaciamiento de los refuerzos. También se considera la reducción de las condiciones posteriores a la COVID-19 de las vacunas actuales, pero la evidencia sobre el alcance de su impacto es inconsistente.
“Actualizado para reflejar que gran parte de la población está vacunada o previamente infectada con COVID-19, o ambos, la hoja de ruta revisada vuelve a enfatizar la importancia de vacunar a quienes aún están en riesgo de enfermedad grave, en su mayoría adultos mayores y aquellos con condiciones subyacentes, incluidos con refuerzos adicionales”, declaró la Dra. Hanna Nohynek, presidenta de SAGE. “Los países deben considerar su contexto específico al decidir si continúan vacunando a grupos de bajo riesgo, como niños y adolescentes sanos, sin comprometer las vacunas de rutina que son tan cruciales para la salud y el bienestar de este grupo de edad”.

EMA RECHAZA LA AUTORIZACIÓN DE COMERCIALIZACIÓN DE MOLNUPIRAVIR
(LAGEVRIO)
24 Febrero de 2023
La Agencia Europea de Medicamentos ha recomendado la denegación de la autorización de comercialización de Lagevrio, un medicamento destinado al tratamiento de la COVID-19 en adultos. La Agencia emitió su dictamen el 23 de febrero de 2023. La empresa que solicitó la autorización, MerckSharp & Dohme B.V., puede solicitar un nuevo examen de la opinión dentro de los 15 días posteriores a la recepción de la opinión.
EL PRAC INICIA UNA REVISIÓN DE LA SEGURIDAD DE LOS MEDICAMENTOS QUE CONTIENEN PSEUDOEFEDRINA
10 Febrero de 2023
Hoy, la Administración de Alimentos y Medicamentos de EE. UU. aprobó la inyección de Tzield (teplizumab-mzwv) para retrasar la aparición de la diabetes tipo 1 en etapa 3 en adultos y pacientes pediátricos de 8 años o más que actualmente tienen diabetes tipo 1 en etapa 2.
La diabetes tipo 1 es una enfermedad que ocurre cuando el sistema inmunitario ataca y destruye las células que producen insulina. Las personas con un diagnóstico de diabetes tipo 1 tienen un aumento de la glucosa que requiere inyecciones de insulina (o el uso de una bomba de insulina) para sobrevivir y deben controlar sus niveles de azúcar en la sangre regularmente durante el día. Aunque puede aparecer a cualquier edad, la diabetes tipo 1 suele diagnosticarse en niños y adultos jóvenes. Una persona tiene mayor riesgo de diabetes tipo 1 si tiene un padre, hermano o hermana con diabetes tipo 1, aunque la mayoría de los pacientes con diabetes tipo 1 no tienen antecedentes familiares.
Tzield se une a ciertas células del sistema inmunitario y retrasa la progresión a la etapa 3 de la diabetes tipo 1. Tzield puede desactivar las células inmunitarias que atacan a las células productoras de insulina, mientras aumenta la proporción de células que ayudan a moderar la respuesta inmunitaria. Tzield se administra mediante infusión intravenosa una vez al día durante 14 días consecutivos.
RECOMENDACIONES DE LA OMS SOBRE VIRUELA SÍMICA Y VACUNA ANTIVARIÓLICA
03 de enero de 2023
En el contexto del Plan Estratégico de Preparación y Respuesta (SPRP) contra la mpox, se presentó al Grupo Asesor Estratégico de Expertos en Inmunización de la OMS (SAGE, por sus siglas en inglés) un borrador de la estrategia de vacunación contra la mpox. Los objetivos de esta estrategia son prevenir la enfermedad humana en grupos con alto riesgo de transmisión, prevenir la propagación a otros grupos, llenar los vacíos de conocimiento para optimizar el impacto en la salud y guiar la acción concertada para mejorar el suministro y el acceso a las vacunas. Se describieron las opciones de acceso, adquisición y regulación de vacunas; las prioridades clave de investigación se destacan dentro de la estrategia.
Se presentó evidencia de una revisión rápida de la literatura sobre la seguridad, inmunogenicidad, eficacia y efectividad de las 3 vacunas contra la viruela actualmente disponibles para su uso en la respuesta al brote. Estas vacunas son MVA-BN (Dinamarca), LC16m8 (Japón) y ACAM2000 (Francia/EE.UU.); las vacunas MVA-BN y LC16m8 han recibido autorización en varios países para su uso en la prevención de la mpox.
Se dispone de datos muy limitados, en su mayoría indirectos, sobre la eficacia de estas vacunas contra la mpox en adultos sanos, y ninguno sobre la protección inducida por la vacuna en poblaciones especiales, como niños, personas inmunodeprimidas y mujeres embarazadas. Del brote actual están surgiendo algunos datos preliminares sobre la eficacia de la vacuna a corto plazo.
LA FDA APRUEBA EL PRIMER MEDICAMENTO QUE AYUDA A REDUCIR LAS REACCIONES ALÉRGICAS A VARIOS ALIMENTOS TRAS UNA EXPOSICIÓN ACCIDENTAL
16 febrero 2024Xolair fue inicialmente aprobado en 2003 para tratar el asma alérgica persistente en ciertos pacientes. También está aprobado para tratar la urticaria crónica espontánea y la rinosinusitis crónica con pólipos nasales en determinados pacientes. Es el primer medicamento aprobado por la FDA para reducir las reacciones alérgicas a más de un tipo de alimento tras una exposición accidental. Es un medicamento (de la clase de los llamados anticuerpos monoclonales) que se une a la inmunoglobulina E (IgE), el tipo de anticuerpo que desencadena las reacciones alérgicas, y bloquea la unión de la IgE a sus receptores.
Los efectos secundarios más comunes de este medicamento incluyen reacciones en el lugar de la inyección y fiebre. Además, el medicamento cuenta con advertencias y precauciones, como la posibilidad de anafilaxia, malignidad y pruebas de laboratorio anormales.